 Hidden sections will not be printed.
Hidden sections will not be printed.General Information
Malondialdehyde (MDA) is a highly reactive compound. Measuring MDA is important in various fields of research such as oxidative stress and ferroptosis. The MDA Assay Kit includes all the necessary components to measure MDA in cellular and tissue samples using the principle below (Figure 1). The kit can be used for both colorimetric and fluorometric assays, depending on the sample type. Compared with other commercially available MDA assay kits, little time is required for assays using this kit (approx. 60 min).
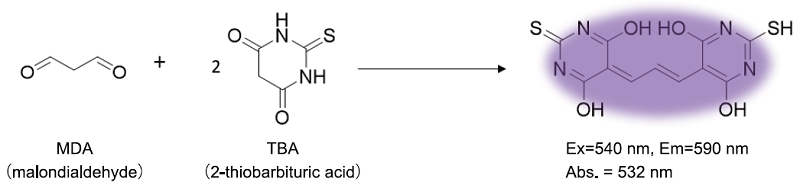
Figure 1. Principle of MDA Assay Kit
Kit Contents
| Lysis Buffer | 6.5 ml x 1 |
| Dilution Buffer | 10 ml x 1 |
| Standard (1 mmol/l) (Blue cap) | 200 μl x 4 |
| Substrate (Red cap) | 58 mg x 1 |
| Antioxidant (Green cap) | 200 μl x 1 |
Storage Conditions
Store at -20°C
Required Equipment and Materials
- Microplate reader (fluorometric measurement: excitation filter 540 nm, emission filter 590 nm, colorimetric: 532 nm filter)
- 96-well plate (fluorometric measurement: black; colorimetric: clear)
- 100–1000 μl, 20–200 μl, 1–10 μl micropipettes
- Phosphate-buffered saline (PBS), dimethyl sulfoxide (DMSO)
- Microtubes
- Conical tubes
- Vortex mixer
- Heating block or water bath (95°C)
- Ice bath
- Homogenizer (only required if tissue samples are used)
Precautions
- Equilibrate reagents to room temperature before use.
- Precipitate may be found in the Lysis Buffer. In this case, dissolve the precipitate by warming the solution. (recommended temperature: 40–50°C).
- Analysis of samples and standards in triplicate is recommended for accuracy.
- Prepare the MDA concentration in samples to be in the range of the standard curve (refer to Table 1).
- One kit enables the measurement of 8 standards and 24 samples in triplicate assays.
Assay method Selection
The fluorometric method is applicable for both cell and tissue samples. For tissue samples, the colorimetric method is an alternative.
Table 1. Selection guide by sample type
| Sample type | Measurement method |
Measurable MDA concentration range |
Sample amount*1 |
| Cell | fluorometric | 1–10 μmol/l | 1–3×107 cells |
| Tissue | fluorometric | 1–10 μmol/l | 10–30 mg |
| colorimetric | 1–50 μmol/l | 20–50 mg |
- Prepare an adequate volume of sample. The sample amounts listed in the Table are the minimum.
- For tissue samples, both the fluorometric method and the colorimetric method can be applicable.
- Allow the fluorometric method to detect MDA with higher sensitivity than the colorimetric method.
Preparation of Solutions
Preparation of Substrate stock solution
Add 400 μl DMSO to the Substrate (red cap) and dissolve using sonication.
- The Substrate stock solution is stable for 2 months when stored at −20°C.
Preparation of Antioxidant PBS solution
Add an appropriate amount of PBS and Antioxidant (green cap) to a tube and mix them to prepare Antioxidant PBS solution (see Table 2 or 3).
- Please be aware that the antioxidant will not dissolve in PBS at this stage (resulting in a cloudy solution). The Antioxidant will be completely dissolved later in the assay procedure during the heating stage of measurement (Measurement step 3 in both fluorometric and colorimetric methods).
- The Antioxidant PBS solution cannot be stored and must be freshly prepared each day.
- Table 2. The required amount of PBS and Antioxidant to prepare Antioxidant PBS solution for fluorometric assay
-
8 samples 16 samples 24 samples PBS 990 μl 1980 μl 2970 μl Antioxidant 10 μl 20 μl 30 μ
- Table 3. The required amount of PBS and Antioxidant to prepare Antioxidant PBS solution for colorimetric assay
-
8 samples 16 samples 24 samples PBS 4950 μl 8910 μl 14850 μl Antioxidant 50 μl 90 μl 150 μl
Preparation of Working solution
- Add Dilution Buffer to a conical tube.
- Add Substrate stock solution to the Dilution Buffer (see Table 4).
- The Working solution cannot be stored and must be freshly prepared each day.
Table 4. The required amount of Working solution by sample number
| Standard (8 serial-dilutions) |
8 samples | 16 samples | 24 samples | |
| Dilution Buffer | 2500 μl | 2500 μl | 4800 μl | 7200 μl |
| Substrate | 90 μl | 90 μl | 172 μl | 260 μl |
General Protocol
Fluorometric Measurement
1. Preparation of MDA standard solutions
- Once opened, Standard cannot be stored. Use an unopened Standard when perform a new experiment on a different day.
- Mix 10 μl Standard (blue cap) and 990 μl double-distilled (dd) H2O in a microtube.
- Serially dilute the solution with dd H2O to prepare standards as shown in Figure 2.
- Transfer 100 μl of the 10 μmol/l standard to a fresh tube (Figure 2).
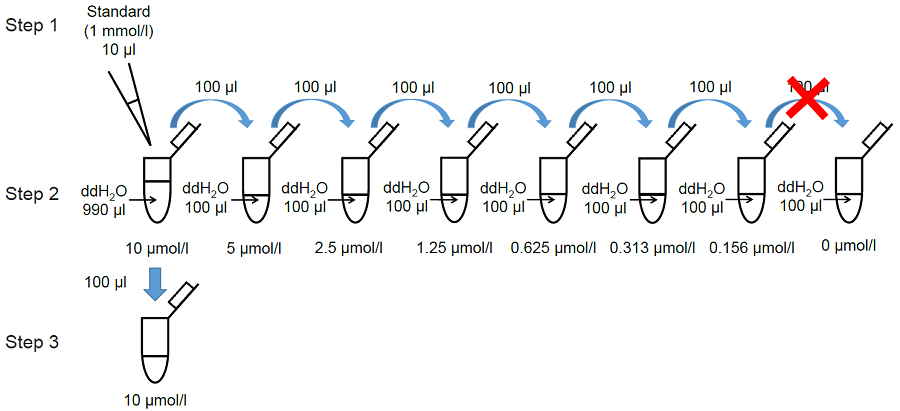
Figure 2. Preparation of MDA standard solutions
2. Sample preparation
-Cell samples-
- Prepare a cell suspension (1–3×107 cells) in a 1.5 ml microtube.
- The suitable cell number may differ depending on the cell line and treatment conditions.
- Centrifuge the cells at 300 × g for 5 min and remove the supernatant.
- Add 1 ml PBS and resuspend the cell pellet by pipetting.
- Centrifuge the cells at 300 × g for 5 min and remove the supernatant.
- Add 100 μl Antioxidant PBS solution and resuspend the cell pellet by pipetting.
-Tissue samples-
- Place the tissue (10–30 mg) in a 1.5 ml microtube.
- Perfuse the tissue sample before the assay as blood components may affect the measurement.
- Optimize the amount of samples for each experiment (see Table 1).
- Add 300 μl Antioxidant PBS solution to the tissue sample.
- Homogenize the sample on ice.
- Make the tissue sample as homogeneous as possible.
- Transfer the homogenized sample to a 1.5 ml microtube and centrifuge at 10000 × g for 5 min.
- Transfer 100 μl of the supernatant to a fresh 1.5 ml microtube
3. Measurement
- Add 100 μl Lysis Buffer to the tube (including a sample or MDA standard solution) and mix using a vortex mixer.
After mixed, incubate the tube for 5 min at room temperature.- Because of high viscosity, the solutions must be mixed by vortexing.
- Normalization of MDA concentration by protein (BCA method) is strongly recommended. In case of high viscosity, take the
viscous sample into and out from a syringe 20–30 times before protein quantification.
- Add 250 μl Working solution to the tube and mix using a vortex mixer.
- Incubate the tube for 15 min at 95°C.
- Cool the tube for 5 min in an ice bath.
- Centrifuge the tube at 10000 × g for 10 min, and transfer 100 μl of supernatant to each well of a 96-well black plate.
- If insoluble material is observed even after the centrifugation, filter (0.22 μm) the supernatant.
- Measure the fluorescence using a microplate reader (recommended filter settings: Ex: 540 nm, Em: 590 nm).
- Calculate the MDA concentration using a calibration curve.
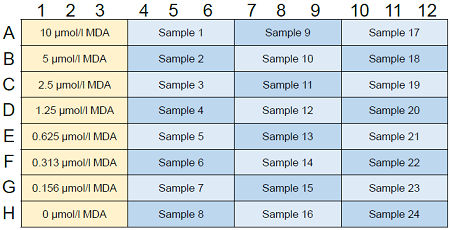
Figure 3. Example of assay plate format (n=3)
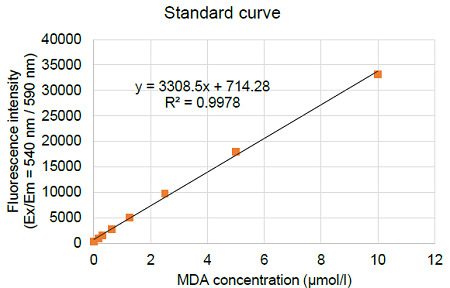
Figure 4. Typical calibration curve generated with MDA standards
Colorimetric Measurement
1. Preparation of MDA standard solutions
- Once opened, Standard cannot be stored. Use an unopened Standard when perform a new experiment on a different day.
- Mix 50 μl Standard and 950 μl double-distilled (dd) H2O in a microtube.
- Serially dilute the solution with dd H2O to prepare standards as shown in Figure 5.
- Transfer 200 μl of the 50 μmol/l standard to a fresh tube (Figure 5).
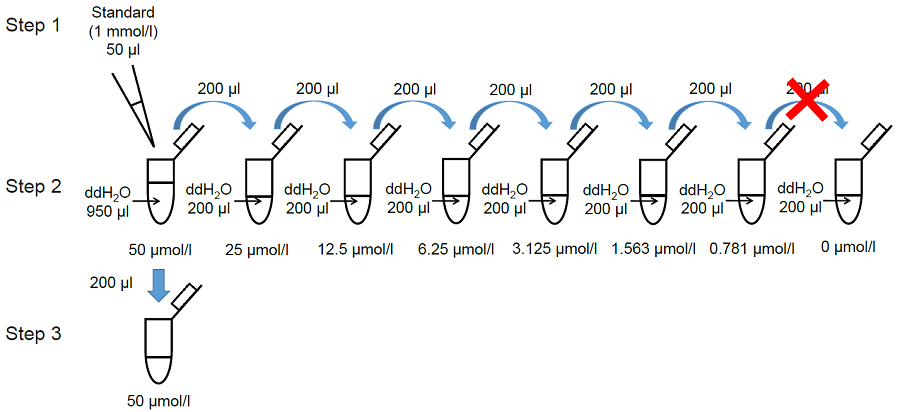
Figure 5. Preparation of MDA standard solutions
2. sample preparation
- Place tissue (20-50 mg) in a 1.5 ml microtube.
- Perfuse the tissue sample before the assay as blood components may affect the measurement.
- Optimize the sample amount for each experiment (see Table 1).
- Add 500 μl Antioxidant PBS solution to the tube.
- Homogenize the sample on ice.
- Transfer the solution prepared in step (3) to a 1.5 ml microtube and centrifuge at 10000 × g for 5 min.
- Transfer 200 μl of the supernatant to a fresh 1.5 ml microtube.
3. Measurement
- Add 200 μl Lysis Buffer to the tube (including a sample or MDA standard solution) and mix using a vortex mixer.
After mixed, incubate the tube for 5 min at room temperature.- Because of high viscosity, the solutions must be mixed by vortexing.
- Normalization of MDA concentration by protein (BCA method) is strongly recommended. In case of high viscosity, take the viscous sample into and out from a syringe 20-30 times before protein quantification.
- Add 300 μl Working solution to the tube and mix using a vortex mixer.
- Incubate the tube for 15 min at 95 °C.
- Cool the tube for 5 min in an ice bath.
- Centrifuge the tube at 10000 × g for 10 min, and transfer 200 μl of supernatant to each well of a 96-well clear plate.
- If insoluble material is observed even after the centrifugation, filter (0.22 μm) the supernatant.
- Measure the absorbance at 532 nm using a microplate reader.
- Calculate the MDA concentration using a calibration curve.
-
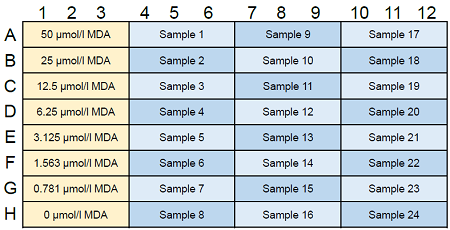
Figure 6. Example of assay plate format (n=3)
-
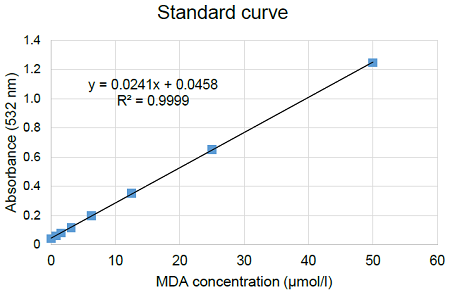
Figure 7. Typical calibration curve generated with MDA standards
Experimental Example
Change in intracellular MDA concentration in erastin -treated HepG2 cells
- HepG2 cells were seeded in a 15 cm dish [1×107 cell/dish in Dulbecco’s modified Eagle’s medium (DMEM) containing 10% fetal bovine serum and 1% penicillin–streptomycin] and cultured overnight in an incubator (37°C, 5% CO2).
- The medium was removed, and erastin solution (100 μmol/l in DMEM) was added to the dish.
- The cells were cultured for 3 h in an incubator (37°C, 5% CO2).
- The medium was removed, and the cells were washed once with 10 ml PBS.
- The cells were trypsinized and collected in a conical tube.
- The erastin-treated HepG2 cells were transferred to a 1.5 ml tube and the cell number was adjusted to 1×107 cells/tube.
- Control cells were transferred to a separate 1.5 ml tube (1×107 cells/tube).
- The supernatants were removed after centrifugation at 300 × g for 5 min.
- PBS (1 ml) was added to each tube, and the supernatants were removed after centrifugation at 300 × g for 5 min.
- Antioxidant PBS solution (100 μl) was added to each tube, and the pellet was resuspended by pipetting.
- Lysis Buffer (100 μl) was added to each tube, and the solution was mixed using a vortex mixer.
- The tubes were incubated at room temperature for 5 min.
- Working solution (250 μl) was added to each tube, and the solution was mixed using a vortex mixer.
- The tubes were incubated at 95°C for 15 min.
- The tubes were cooled on ice for 5 min.
- The supernatant (100 μl) was transferred to each well of a 96-well black plate after centrifugation at 10000 × g for 10 min.
- Fluorescence was measured using a microplate reader (Ex: 540 nm, Em: 590 nm), and the concentration of MDA in the samples was calculated from the calibration curve.
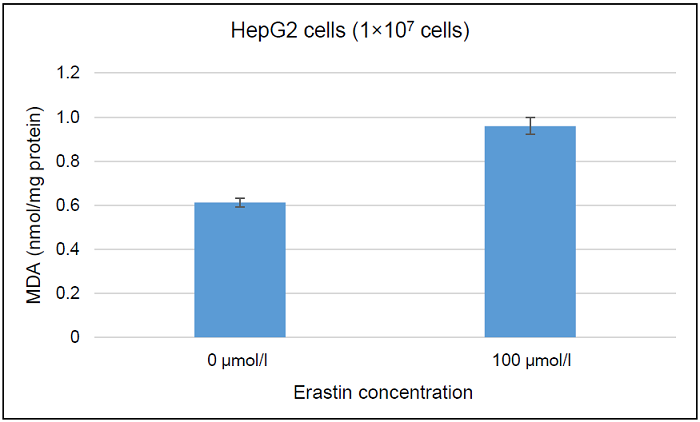
Figure 8. Determination of intracellular MDA concentration in erastin-treated and non-treated HepG2 cells
Measurement of MDA concentration in KK-Ay mouse liver
- Antioxidant PBS solution (500 μl) was added to a 1.5 ml microtube containing the tissue sample (30 mg).
- The tissue sample was homogenized using a Dounce homogenizer.
- The solution prepared in step (2) was transferred to a 1.5 ml microtube.
- The supernatant (200 μl) was transferred to a fresh 1.5 ml microtube after centrifugation at 10000 × g for 5 min.
- Lysis Buffer (200 μl) was added to the tube, and the solution was mixed using a vortex mixer.
- The tube was incubated at room temperature for 5 min.
- Working solution (300 μl) was added to the tube, and the solution was mixed using a vortex mixer.
- The tube was incubated at 95°C for 15 min.
- The tube was cooled on ice for 5 min.
- The supernatant (200 μl) was transferred to each well of a 96-well clear plate after centrifugation at 10000 × g for 10 min.
- Absorbance at 532 nm was measured using a microplate reader, and the concentration of MDA in the samples was calculated from the calibration curve.
Table 5. MDA concentration in KK-Ay mouse liver
| MDA concentration (µmol/l) | |
| KK-Ay mouse liver (30 mg) |
21.15 |
Frequently Asked Questions / Reference



M496: MDA Assay Kit
Revised Mar., 18, 2025